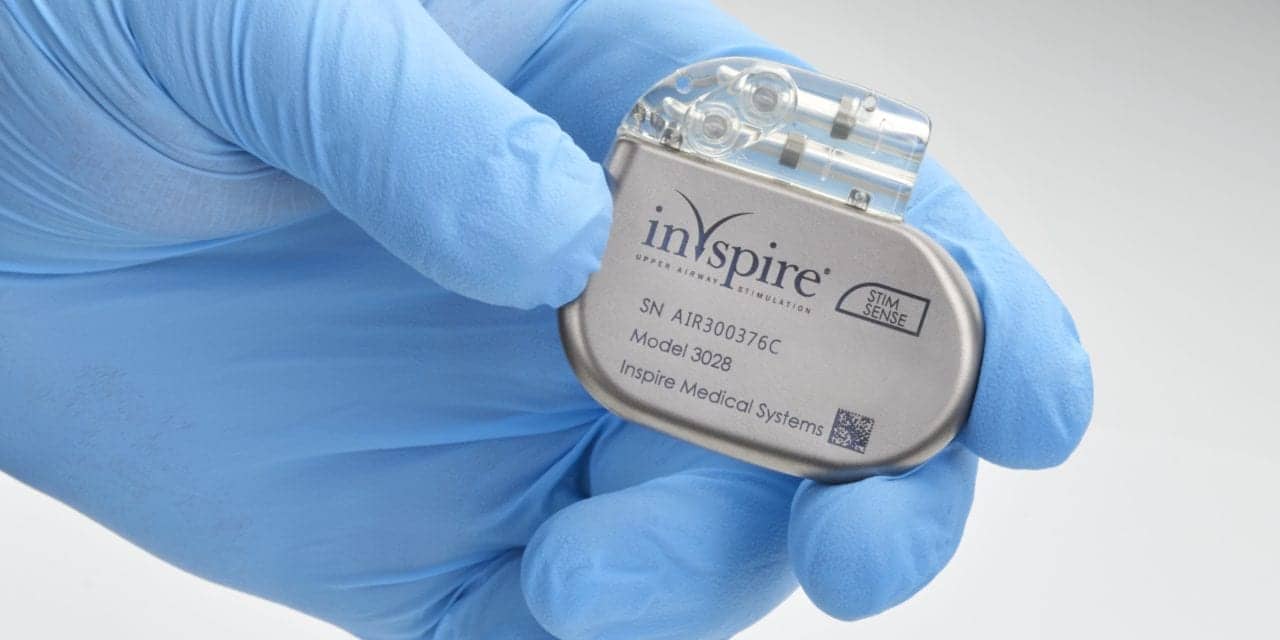
The recall addresses a defect in a subset of Inspire IV implantable pulse generators that may cause malfunctions and necessitate revision surgery for patients.
Summary: Inspire Medical Systems has issued a recall impacting 32 of its Inspire IV implantable pulse generators used in treating obstructive sleep apnea due to a manufacturing defect. This defect may cause system malfunctions, including electrical leakage, potentially leading to serious health consequences and necessitating revision surgery. The FDA has classified this as the most serious type of recall.
Three Key Takeaways:
- Defect and Recall: Inspire Medical Systems issued a field corrective action for 32 Inspire IV implantable pulse generators due to a manufacturing defect that can cause electrical malfunctions, leading to potential serious health risks.
- Patient and Provider Actions: Affected patients are advised to contact their healthcare providers for routine check-ups and possible revision surgery, while providers are urged to notify patients and monitor the devices closely.
- FDA Classification: The FDA has identified this recall as the most serious type.
Inspire Medical Systems is recalling Inspire IV implantable pulse generator model 3028, a key component of the Inspire upper airway stimulation system for treating obstructive sleep apnea, due to a manufacturing defect.
A recall in this context means taking corrective actions, such as informing patients and healthcare providers, or conducting additional monitoring. According to an Inspire Medical spokesperson, the primary goal is to ensure patient safety without requiring device removal or revision unless necessary. This approach helps manage risks while minimizing unnecessary procedures.
This defect can cause system malfunctions after implantation, leading to electrical leakage in the sensing circuit. As a result, patients may need revision surgery to replace the implantable pulse generator and restore therapy.
The use of the affected product may cause serious adverse health consequences, including stimulation below normal therapeutic levels and/or early depletion of the battery (resulting in loss of therapy), inappropriate or inconsistent stimulation effect, painful stimulation, or perceived shocking sensation and death, according to a medical device recall notice from the US Food and Drug Administration (FDA).
“Patient safety has always been, and continues to be, of paramount importance to Inspire. To that end, we have issued a voluntary field corrective action of all 24 potentially impacted units,” says Phil Ebeling, chief operating officer at Inspire Medical. According to Ebeling, 24 patients are notified as part of this action, while eight have been medically managed, for a total of 32 impacted patients.
There have been no reported injuries or death deaths, according to the FDA’s medical device recall notice.
Ebeling notes that Inspire Medical has notified regulatory authorities, including the FDA, and sent a letter to impacted health care providers “with specific instructions, including notifying the patients of the recall and requesting that they come into the clinic for testing to ensure that the units are operating properly.”
“We have also advised health care providers to continue monitoring their patients to make sure that the units continue to operate properly and consistently throughout the course of therapy,” says Ebeling.
The implantable pulse generator stores therapy settings configured by a physician and delivers mild electrical stimulation to the hypoglossal nerve, which controls tongue muscles, to maintain airway patency during sleep. The implantable pulse generator works together with external programmers that allow the physician to set and adjust the therapy parameters and the patient to control the therapy’s activation and intensity.
This recall advises doing regular check-ups and tests to analyze signals and resistance at every visit. According to the Inspire spokesperson, these simple tests can spot issues with the device without needing surgery.
The FDA has identified this recall as a class 1 recall, most serious type. This device may cause serious injury or death.
Affected Product
- Product names: Inspire IV implantable pulse generator
- Unique device identifier/Model: 0855728005915/Model 3028
- Lot/serial numbers: Includes 32 devices of model 3028 implantable pulse generator. See full list of affected devices.
What to Do
On June 17, Inspire Medical sent all affected customers an Urgent Medical Device Correction recommending the following actions:
Healthcare Providers:
- Notify affected patients of this voluntary recall.
- Schedule an appointment for the patient to check if their Inspire therapy is working properly by analyzing signals and resistance. Keep an eye out for any changes in the stimulation, lack of therapy effectiveness, or problems with turning the therapy on.
- Keep doing regular check-ups and tests to analyze signals and resistance at every visit, as these simple tests can spot issues with the device without needing surgery.
Patients:
- Contact your healthcare provider as soon as possible to make sure you have a routine office visit scheduled.
- If you have already been contacted by your healthcare provider, follow up as scheduled.
- Attend your scheduled office visit. Routine non-invasive diagnostic monitoring identifies this potential implantable pulse generator defect.
- If you have new symptoms or re-occurrence of symptoms like fatigue, perceived sleepiness, snoring problems, etc, contact your sleep physician for a comprehensive evaluation, which may include a polysomnography.
- If your provider determines a revision surgery is necessary to replace the implantable pulse generator, you may contact Inspire’s Patient and Physician Services at 1-844-OSA-HELP (1-844-672-4357) for further information.
“Please rest assured that we will continue to cooperate fully and follow up, as appropriate, with the appropriate regulatory authorities, including the FDA, health care providers, and all potentially impacted patients,” says Ebeling.
Health care professionals and consumers may report adverse reactions or quality problems they experienced using these devices to MedWatch: The FDA Safety Information and Adverse Event Reporting Program.
7/9/24: Article updated with information from Inspire Medical





{kind=link}