
Unexpected electrical status epilepticus of sleep (ESES) in a routine pediatric polysomnogram
A 5-year-old girl with microcephaly was initially referred to the University of New Mexico Sleep Disorders Center for evaluation of suspected pediatric obstructive sleep apnea. Following adenotonsillectomy, the patient’s postoperative polysomnogram (PSG) results showed almost continuous bilateral generalized spike-wave discharges in NREM sleep. Her case illustrates that sleep clinicians and technologists need to recognize the distinctive age-related electroencephalogram (EEG) characteristic of electrical status epilepticus of sleep (ESES), understand its clinical significance and morbidity, and the need for a prompt neurological referral.
BACKGROUND
ESES is an EEG pattern characterized by continuous (or near continuous) spike-wave discharges during NREM sleep. It is often associated with an encephalopathy (ESES syndrome) characterized by epileptic seizures, continuous spike-wave discharges in NREM sleep, global or selective regression of cognitive functions, and motor impairments. The pathophysiological basis of ESES is poorly understood.
Beenhakker and Huguenard (2009) have suggested that the ESES pattern may represent hyperexcitable neuronal firing of the same corticothalamic neuronal network that generates sleep spindles in NREM sleep. ESES may occur in children with no previous neurologic problems (idiopathic type) as well as in children with preexisting neurodevelopmental impairment and/or marked structural changes in the nervous system (symptomatic form). The EEG pattern and seizures of ESES appear somewhat abruptly usually between ages 5 and 9 years and disappear by puberty, but permanent neuropsychological sequelae remain in half the patients. Certain antiepileptic medications may aggravate seizures and ESES while other medical and/or epilepsy surgeries abolish the EEG pattern often then accompanied by cognitive catch-up. Earlier recognition and treatment of the EEG pattern and clinical syndrome may lead to better outcomes.
PATIENT CASE
A 5-year-old girl with microcephaly was initially referred to the University of New Mexico Sleep Disorders Center for evaluation of suspected pediatric obstructive sleep apnea. Her symptoms of loud nightly snoring, waking, and daytime tiredness and hyperactivity, as well as 3+ tonsillar hypertrophy were consistent with the suspected diagnosis. Her birth and medical history, and routine physical examination, were unremarkable. A diagnostic PSG performed on December 13, 2010 demonstrated moderate-severe pediatric obstructive sleep apnea with a pediatric obstructive apnea-hypopnea index (pediatric OAHI) of 8.5 events per hour of sleep (12 events per hour of sleep in the supine position) and an oxygen saturation nadir of 85%. No abnormalities were noted in the limited EEG channels recorded. Due to her clinical symptoms and polysomnogram results, she was referred to a pediatric otolaryngologist (ENT) and underwent an adenotonsillectomy. Following surgery, her parents reported she was sleeping better, faint snoring remained, daytime sleepiness had resolved, and hyperactive behavior lessened.
A postoperative PSG was performed on August 20, 2011. The study was reassuring for an absence of sleep-disordered breathing; however, almost continuous bilateral generalized spike-wave discharges were observed in NREM sleep (Figure 1). This prompted us to carefully review the entire PSG in wake and REM sleep. We found only rare interictal epileptic discharges in REM sleep (Figure 3) and none awake (Figure 2). We referred the child to a pediatric neurologist specializing in epilepsy for further evaluation and aggressive intervention.
DISCUSSION
ESES syndrome is a rare age-related condition occurring only in childhood with a suggested incidence of 0.2% to 0.5% of childhood epilepsies. ESES consists of sleep-induced continuous paroxysmal discharges of spike-wave complexes with a frequency of 1.5 to 3.5 Hz (cycles per second) on the EEG. ESES may be continuous or discontinuous during sleep and is usually diffuse and bilateral.
POLYSOMNOGRAM EEG FINDINGS
The EEG pattern of ESES is characterized by the appearance of continuous (or near continuous) generalized spike-wave discharges during NREM sleep, which typically appear as soon as the child falls asleep. The discharges in NREM sleep are typically generalized and symmetrical, and typically repeat at frequencies of 1.5 to 2 per second. However, discharges in some during NREM sleep may be asymmetrical, lateralized, or relatively focal, often with maximal negativity frontally or centrally, and sometimes consisting of sharp rather spike-like discharges. Discharges in wakefulness are infrequent, focal, or multifocal spike or spike waves. During REM sleep, spike waves occur less frequently, occupying less than 25% of the REM sleep time. Discharges during REM are often focal and similar to those in the wake EEG. A typical EEG finding in these patients before ESES develops is focal spikes or spike waves predominantly in the centrotemporal or frontotemporal regions. The ESES pattern typically normalizes 3 to 5 years after the onset of the syndrome, sooner with effective medical and/or epilepsy surgery therapies. At first glance, the ESES pattern is typically generalized, but ESES is a focal epilepsy: the discharges generalize during NREM sleep, and are focal (or multifocal) during wake and REM sleep.
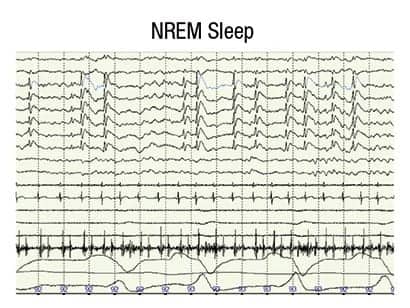
Figure 1. Generalized bilateral 1-2 per second spike and slow wave complexes that at times occurred in continuous runs and persisted into NREM II and NREM III sleep.
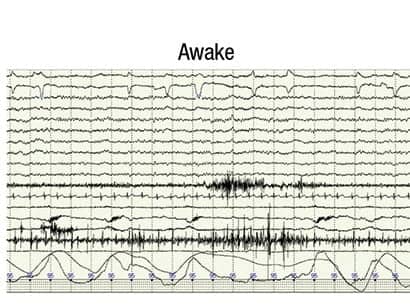
Figure 2. Complete resolution of discharges.
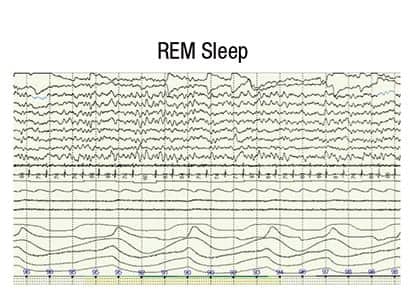
Figure 3. Interictal discharges occur infrequently in REM sleep due to the desynchronized EEG pattern of REM sleep.
SLEEP ARCHITECTURE
Sleep spindles, K-complexes, and vertex waves are seldom recognizable during ESES. The percentage of continuous spiking during NREM sleep often lessens across sleep cycles (often >90% in the first cycles of NREM sleep, averaging >85% for all NREM sleep cycles), and later NREM periods contain lower percentages of spike-wave discharges. REM sleep is often recognized by disappearance of the ESES pattern (Figure 3). With treatment and time, the continuous spiking becomes less, permitting sleep stage scoring. When the ESES pattern is less continuous, sleep architecture seems preserved with approximately 80% of total sleep time spent in NREM, 20% in REM sleep. Sleep organization and sleep stages are normal after ESES disappears. During wakefulness, the EEG may be normal or mildly abnormal showing paroxysmal foci in the frontotemporal or centrotemporal regions or a brief burst of more diffuse spike-wave activity, but in slow-wave sleep discharges become continuous. Epileptiform activity may cause physical, cognitive, and behavioral impairments. Even brief discharges of a few seconds, without clearly perceptible clinical symptoms, can cause cognitive dysfunction, so-called transient cognitive impairment.
ESES was first reported by Patry and colleagues in 1971, and they called the EEG pattern electrical or subclinical status epilepticus because it was not associated with any clinical signs when it occurred during sleep. All the children had moderate to severe cognitive impairment, and five had epileptic seizures when the ESES pattern was first observed. ESES is an epileptic encephalopathy with the onset most often associated with or followed by neurologic regression and seizures. The International League Against Epilepsy (ILAE) calls it “epileptic encephalopathy with continuous spike and wave during sleep (CSWS).” The ILAE classification defines the ESES syndrome as an age-related and self-limited disorder characterized by (1) epilepsy with focal and/or generalized seizures; (2) neuropsychological impairment in the form of global or selective regression of cognitive function; (3) motor impairment in the form of ataxia, dyspraxia, dystonia, or unilateral deficit; and (4) typical EEG findings with a pattern of diffuse spike waves (unilateral or focal occurring in up to 85% of slow-wave sleep and persisting on three or more recordings over a period of at least 1 month).
The first clinical symptoms typically occur between 5 and 9 years of age. The first seizure occurs in sleep in half of the cases, and in 40% it consists of unilateral convulsions that can last more than 30 minutes (hemiclonic status epilepticus). Although other seizure types may be observed, tonic seizures are never seen in patients with ESES.
The second stage is often observed 1 to 2 years after epilepsy onset with the development of ESES with specific or global neuropsychological deterioration or behavioral problems. Habitual seizures become more frequent, and new seizure types are often observed such as hemifacial or hemiconvulsive. Convulsive seizures continue to occur primarily during sleep. The neuropsychological regression tends to be gradual and progressive. Other neurologic deficits that occur in the second stage include ataxia, hemiparesis, or planning of movements and coordination. Some develop dysarthria, drooling, face/tongue weakness, or speech arrest.
The third stage of ESES is a residual stage with the disappearance of the epilepsy and ESES by puberty; however, permanent neuropsychological sequelae remain in 50% of patients.
Landau-Kleffner syndrome (LKS) has similar characteristics to ESES and was first described in 1957 in six children with convulsive disorder and an acquired aphasia. LKS affects the parts of the brain that control comprehension and speech (Broca’s area and Wernicke’s area). LKS is a severe, partly reversible, age-related childhood clinical syndrome, characterized by verbal auditory agnosia, quickly followed by regression of spontaneous speech, and in three of four cases seizures and behavioral disorders.
An acquired auditory agnosia is seen that may progress over weeks to months, along with spike-wave abnormalities on EEG during sleep. In most of the children, language and development are normal before the onset of LKS. Some children have symptoms including irritability, attention deficit disorder, and autistic-like behavior disorder. Children may be unable to understand spoken language or appear deaf.
Seizures are typically infrequent and easily treated. Most common seizure types include generalized clonic, partial clonic, and atypical absence seizures. Atonic seizures are not frequently seen.
TREATMENT
Treatment choices for ESES syndrome are based primarily on case reports and small case series. Various antiepileptic drugs, benzodiazepines, sulthiame, corticosteroids, and intravenous immunoglobulin (IV-IG) have been reported to abolish the ESES pattern from the EEG. Agents such as oxcarbazepine, phenobarbital, levetiracetam, clobazam, and carbamazepine have been shown to aggravate seizures and should be avoided. Regardless of the intervention, long-term remission rates have been reported at best, up to 45%. Better results have been shown in those who underwent epilepsy surgery. Poorer outcomes have been shown in patients who developed ESES at a younger age, and children with a higher IQ at the time of ESES diagnosis had better cognitive outcomes. Complete neuropsychological and cognitive recovery is possible but rarely observed.
ESES demonstrates that it is imperative that every pediatric sleep specialist be suspicious of any EEG interictal rhythm that commences with the onset of NREM sleep. Although the etiology may not be readily apparent, prompt referral to a neurologist is mandatory.
Shahana Masood, MD, completed a sleep medicine fellowship at the University of New Mexico and has completed fellowships in pulmonary and critical care medicine. Frank M. Ralls, MD, is an assistant professor of internal medicine and program director for Sleep Medicine at the University of New Mexico. Corresponding author Madeleine M. Grigg-Damberger, MD, is professor of neurology, University of New Mexico School of Medicine.
Recommended Reading
1. Tassinari CA, Rubboli G, Volpi L, et al. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin Neurophysiol. 2000;111 Suppl 2:S94-S102.
2. Zhang J, Talley G, Kornegay AL, Edwards JC. Electrical status epilepticus during sleep: a case report and review of the literature. Am J Electroneurodiagnostic Technol. 2010;50(3):211-218.
3. Tovia E, Goldberg-Stern H, Ben Zeev B, et al. The prevalence of atypical presentations and comorbidities of benign childhood epilepsy with centrotemporal spikes. Epilepsia. 2011;52(8):1483-1488.
4. Su XM, Zhang GP, Yang BZ, Li L. [Clinical analysis of 4 cases of childhood Rolandic epilepsy with electrical status epilepticus during sleep]. Zhongguo Dang Dai Er Ke Za Zhi. 2011;13(4):344-345.
5. Saadeldin IY, Al-Tala SM. Coexistence of epileptic encephalopathy with continuous spike-and-wave during sleep, atypical benign partial epilepsy, and fixation-off sensitivity in two siblings. Epilepsy Behav. 2011;20(1):116-122.
6. Peltola ME, Liukkonen E, Granstrom ML, et al. The effect of surgery in encephalopathy with electrical status epilepticus during sleep. Epilepsia. 2011;52(3):602-609.
7. Loddenkemper T, Fernandez IS, Peters JM. Continuous spike and waves during sleep and electrical status epilepticus in sleep. J Clin Neurophysiol. 2011;28(2):154-164.
8. Hughes JR. A review of the relationships between Landau-Kleffner syndrome, electrical status epilepticus during sleep, and continuous spike-waves during sleep. Epilepsy Behav. 2011;20(2):247-253.
9. Bolsterli BK, Schmitt B, Bast T, et al. Impaired slow wave sleep downscaling in encephalopathy with status epilepticus during sleep (ESES). Clin Neurophysiol. 2011;122(9):1779-1787.
10. Overvliet GM, Besseling RM, Vles JS, et al. Nocturnal epileptiform EEG discharges, nocturnal epileptic seizures, and language impairments in children: review of the literature. Epilepsy Behav. 2010;19(4):550-558.
11. Liukkonen E, Kantola-Sorsa E, Paetau R, Gaily E, Peltola M, Granstrom ML. Long-term outcome of 32 children with encephalopathy with status epilepticus during sleep, or ESES syndrome. Epilepsia. 2010;51(10):2023-2032.
12. Garcia-Penas JJ. [Neurocognitive dysfunction in electrical status epilepticus during slow-wave sleep syndrome: can the natural course of the syndrome be modified with early pharmacological treatment?]. Rev Neurol. 2010;50 Suppl 3:S37-47.
13. Tuchman R. CSWS-related autistic regression versus autistic regression without CSWS. Epilepsia. 2009;50 Suppl 7:18-20.
14. Scheltens-de Boer M. Guidelines for EEG in encephalopathy related to ESES/CSWS in children. Epilepsia. 2009;50 Suppl 7:13-17.
15. Kramer U, Sagi L, Goldberg-Stern H, Zelnik N, Nissenkorn A, Ben-Zeev B. Clinical spectrum and medical treatment of children with electrical status epilepticus in sleep (ESES). Epilepsia. 2009;50(6):1517-1524.
16. Wang SB, Weng WC, Fan PC, Lee WT. Levetiracetam in continuous spike waves during slow-wave sleep syndrome. Pediatr Neurol. 2008;39(2):85-90.
17. Nickels K, Wirrell E. Electrical status epilepticus in sleep. Semin Pediatr Neurol. 2008;15(2):50-60.





{kind=link}